周期4周左右,具体周期根据企业产品调整
服务一对一专业服务
其他认证服务IVDR CE/FDA 510K/FDA 验厂/UKCA/CFS/产品检测等
认证内容MDR CE服务
服务范围全国
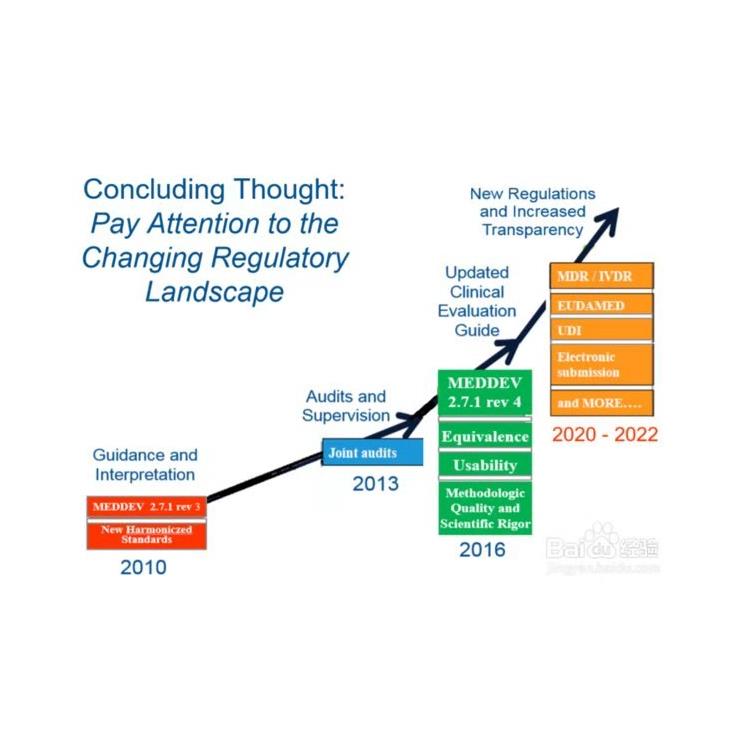
新的欧盟医疗器械法规(MDR CE)将对在欧盟内开展贸易的所有制造商的标签流程产生重大影响。新法规影响深远,标签流程也因此成了公司的头等大事。标签标注不清晰、不符合MDR要求都有可能造成公司无法继续在欧洲销售其产品,从而造成大量库存积压,甚至引发成本高且损害公司声誉的产品召回事件。
欧盟新规申报难度**FDA,强制公告机构(NB)门槛提高:
在过去,欧盟CE认证的难度较中国NMPA及美国FDA更低,背后的原因则是欧盟医疗器械旧法规的约束力松弛,对医疗器械研发商报批要求普遍较低。同时也导致了部分仅获得CE认证的医疗器械在欧盟地区落地后也常出现医疗事故,所以这些产品未来进入中国及美国市场时,仍然需要面临更长期、更严格的报批流程。
在新规执行之前,一些低风险医疗器械产品研发厂商在申请CE认证时,可以通过自我声明的方式进行申请,但这种申请的方式监察并不严格、缺乏约束力。
如果产品没办法准确管控,那么CE认证的“含水量”便较高。针对欧盟这种情形,如果你的产品只在欧盟拿了认证,那么该产品在英德申报时还是需要经历严格的临床试验,以确保产品的*性、有效性。

MDR CE对于产品标签的要求:
1、器械的名称或商品名称;
2、使用者识别器械所必需的详细信息、包装内容以及对于使用者不明显的器械预期用途;
3、制造商的名称、注册商号或注册商标及其注册营业地点的地址;
4、授权代表的姓名和授权代表的注册营业地点地址(若制造商在欧盟以外有其注册营业地点);
5、若没有指明可*使用的日期,则指明制造日期。若日期清晰可辨,制造日期可作为批号或序列号的一部分;
6、指明适用的任何特别储存和/或处理条件;
7、若以无菌方式提供器械,还应指示其无菌状态方法;
8、需要立即引起器械使用者和任何其他人的注意、需要采取的警戒或预防措施。

在欧盟MDR新规执行之后,即便是一些电商平台上进行销售的医用产品也需要通过严格的授权公告机构(Notified Body,NB/欧代)进行申报。与此同时,欧盟对NB机构的要求也大幅提升,公告机构是单个于进行符合性评估活动的产品制造商的第三方机构,要求长期配备具有相关证书/资质的产品审查人员、质量管理体系审核人员等,且不能采取外包机制聘用。对公告机构(NB)的高要求,也让目前获批的能够进行新规公告的机构数量大幅降低。

从Directive(指令)到Regulation(法规),欧盟提高了对医疗器械的约束力,发布立即在欧盟成员国生效并成为有约束力的法律,此次的Regulation*像Directive那样需要经过成员国转化成当地法律法规再去落地实施。
值得一提的是,在欧盟医疗器械分类当中,将医疗器械划分为医疗器械(MD)和体外诊断器械(IVD)两大类,目前受到新规MDR管辖的限于MedicalDevices,体外诊断器械的相应新规IVDR的执行时间为2022年5月26日,也意味着IVD器械厂商还有一年的时间可以缓冲和适应新规变化,并做好应对准备工作。
我们公司以客户的满意度为标准,专注细节,坚守承诺,快速行动!用现代化的企业管理制度打造出一支优良的员工队伍,可为客户提供个性化的服务。
http://sungo13760748565.b2b168.com